I due prodotti sono ad uso ospedaliero.
Ai sensi dell’art. 84 comma 6. non possono essere ne detenuti in scorta zooiatrica ne prescritti. Tali prodotti infatti sono utilizzabili solo all’interno delle strutture veterinarie.
Farmaco - FAQ
Per sottoporre un quesito a FNOVI si raccomanda di utilizzare il form della sezione contatti selezionando "FARMACO" come oggetto della richiesta.
Nei quesiti devono essere indicati nome e cognome (che non saranno pubblicati) e numero di iscrizione all'Ordine dei medici veterinari o dei farmacisti.
Domande e risposte saranno pubblicate in questa sezione e al mittente sarà inviata una mail che avvisa della pubblicazione.
Si precisa che per quanto attiene ai documenti citati (leggi, note e circolari ministeriali, foglietti illustrativi, siti internet o altre fonti) il loro contenuto è riferito al momento della risposta, se non altrimenti indicato.
Per la ricetta elettronica veterinaria si rammenta che sulla piattaforma è disponibile un corso FAD gratuito e accreditato ECM.
Domanda nr. 320
Inserita il 09/04/2014
Domanda:
E' possibile per un medico veterinario dotato di scorta zooiatrica dotarsi di una sola confezione di FENTANEST® FIALE o REMIFENTANIL® FIALE (entrambi farmaci umani, non è presente il farmaco veterinario da utilizzare in un piccolo animale? Anche una singola confezione viene definita scorta?
Risposta:
Domanda nr. 319
Inserita il 09/04/2014
Domanda:
Riguardo alla segnalazione di farmacovigilanza in alcune aziende di bovine da latte ci sono più farmaci che vengono utilizzati in uso improprio ovvero per un tempo maggiore all'AIC e con un dosaggio a volte superiore. Io posso effettuare una o più segnalazioni a seconda che vi siano 3 o più molecole da indicare, dove scrivo "assenza di efficacia attesa" e una volta effettuata questa segnalazione utilizzare quelle molecole in uso improprio? Sarebbe molto complicato infatti effettuare le segnalazioni per ogni animale trattato in uso improprio.
Risposta:
La domanda, che per la descrizione della condizione sembra essere riferita all'uso degli antibiotici, pone diversi quesiti assieme.
Per quanto attiene alla identificazione degli animali la scheda non richiede l'identificazione singola degli animali trattati in gruppo. Sarà dunque il veterinario ad individuare di volta in volta se sia opportuno individuare gli animali (es livelli di reazioni diverse e dunque osservazioni e trattamenti conseguenti diversi a parità di prodotto somministrato)
Per quanto riguarda l'utilizzazione di 3 molecole, se queste sono utilizzate contemporaneamente la scheda prevede che venga indicato in modo da consentire lo studio sulle cause della diminuzione di efficacia che potrebbero essere dovute ad interazioni tra farmaci.
La segnalazione di farmacovigilanza è uno degli elementi che consentono il proseguimento della terapia in uso improprio ma non può essere una giustificazione a tale uso.
Il medico veterinario dovrà in ogni caso nel tempo supportare la sua scelta terapeutica con antibiogrammi o con altri elementi diagnostici utili al fine di definire le motivazioni di tali scelte terapeutiche dal momento che il farmaco viene utilizzato in modalità differenti rispetto a quanto è scritto sul foglietto illustrativo.
Per quanto attiene alla ripetizione della terapia in soggetti diversi per la stessa patologia, a rigor di legge ogni uso improprio richiederebbe una segnalazione.
A supporto di un comportamento diverso, che comunque è bene ricordarlo non è in ottemperanza alla legge, possono tuttavia trovare riscontro antibiogrammi periodici, protocolli operativi, azioni di miglioramento in temi di biosicurezza documentati e quant'altro dimostri l'impegno al controllo e contenimento dell'uso degli antibiotici.
Domanda nr. 318
Inserita il 09/04/2014
Domanda:
Sono un farmacista della provincia di Verona. Ho ricevuto una richiesta di Temgesic® fiale e Ipnovel® fiale da parte di un ambulatorio veterinario: la ricetta fornitami nonera il modello ministeriale per gli stupefacenti e tanto meno quello a ricalco destinato per animali da macello. Semplicemente 3 fogli formalmente corretti. A quanto mi risulta per gli stupefacenti della Tab II sez. A (proprio Temgesic® ) anche per scorte serve la prescrizione su ricettario ministeriale.
Risposta:
Le tabelle a cui fare riferimento sono previste dall'art. 14 del DM 309/90. Il Temgesic® fiale è in tab IIA per cui ci vuole la richiesta in triplice copia su carta intestata dell'ambulatorio come previsto dall'art. 42 del DPR 309/1990.
L'Ipnovel® è un ospedaliero in Tab IID per cui per l'approvvigionamento per scorta ci vuole la RNRTC, prevista in all. III del D. Leg. 193/2006.
Domanda nr. 317
Inserita il 09/04/2014
Domanda:
Se si prescrive con RNRTC a un canile rifugio municipale, gestito da una associazione, un quantitativo esagerato di flaconi di antibiotico da 250 ml anche per animali DPA(bovini-ovini-suini-cani-gatti) e diverse confezioni di Noromectin® per bovini/suini e non viene sulla ricetta identificato a quali e quanti animali deve essere fatta la terapia e per quanto tempo,ne se trattasi di scorte proprie o dell'impianto e tenendo presente che il canile non è autorizzato alla detenzione delle scorte e non è in possesso del registro dei trattamenti ; si viene a chiedere: se non vi è evidente traccia, sull'uso che è stato fatto, di quei farmaci nel canile, in quanto non più presenti; a)quali responsabilità , se del caso, sono in capo al veterinario che ha redatto quella ricetta totalmente incompleta; b)a quali responsabilità e/o sanzioni (se del caso) sono in capo al veterinario responsabile del canile (responsabile sanitario, che comunque non ha prescritto quella ricetta redatta da altro veterinario) e al responsabile che gestisce il canile?
Risposta:
A raffronto di una situazione che sia per l’uso in deroga difficilmente giustificabile in una percentuale così alta di casistiche, sia per le quantità prescritte, descrive l’evidenza di un utilizzo a sproposito del farmaco veterinario, la fattispecie descritta non trova applicabile nemmeno una sanzione.
Infatti non esiste la sanzione per l’errata compilazione della ricetta, non esiste la sanzione per chi, non avendo la scorta, non è tenuto a scaricare i farmaci in confezioni autorizzate anche o esclusivamente per animali destinati alla produzione di alimenti per l'uomo, non esiste la sanzione per il mancato possesso della scorta.
Difficile anche l’applicazione della sanzione relativa all’articolo 10 cercando di capire perché sia stato usato il Noromectin® invece del Filarive® in compresse o altro farmaco antiparassitario registrato per cani in assenza di una diagnosi scritta e documentata che possa confutare l’uso in deroga.
Domanda nr. 316
Inserita il 07/04/2014
Domanda:
L'utente finale (proprietario/detentore) di un cavallo NONDPA per quanto tempo deve conservare la copia gialla della RNRTC? E' giusto se a fine trattamento (senza quantità residuali), il proprietario dell'equide getta la ricetta?
Risposta:
Come indicato dalla nota Ministeriale 15952/2007, p.to 6 ultimo paragrafo il comportamento descritto è corretto.
Domanda nr. 315
Inserita il 01/04/2014
Domanda:
In merito alla FAQ n° 10, in un allevamento senza scorte si trova la RNRT per un trattamento antibiotico di massa avvenuto pochi giorni prima, ma non ancora registrato nel registro dei trattamenti, nè dall'allevatore, nè dal medico veterinario. Ora l’art. 15 comma 2 del DLgs 158/06 prevede che l’allevatore debba registrare il trattamento farmacologico entro 24 ore dall'inizio e dalla fine ma da nessuna parte la legge indica i tempi entro i quali lo debba fare il medico veterinario. Men che meno viene indicato l’obbligo della contestualità come indicato in quella FAQ. E’ esatto?
Risposta:
In effetti gli obblighi del veterinario inquanto a tempi di trascrizione non sono indicati nella legge. L’indicazione dei tempi è indicata solo nelle LINEE GUIDA APPLICATIVA del Decreto Legislativo del 16 marzo 2006, n. 158 trasmesse con nota 7835 del 4 marzo 2013. Tale nota detta prescrizioni di legge che pur risolvendo una carenza della legge non ne ha la forza impositiva.
Infatti, tutte le volte che una normativa europea rimanda all'atto di recepimento per dettare termini e sanzioni, specifica che ciò debba avvenire per atto di recepimento, cosa che una nota ministeriale non è mai.
Si ringrazia l’avv. Scarciglia per la consulenza.
Domanda nr. 314
Inserita il 31/03/2014
Domanda:
In relazione a quanto sotto riportato, Vi chiedo cosa si intenda per ultima registrazione riferita sia al registro trattamenti sia alle ricette unitamente alla documentazione commerciale. "Art. 79 (Registro dei trattamenti di animali destinati alla produzione di alimenti). - 1.(...) 2. Il registro di cui al comma 1, a pagine prenumerate e vidimato dalla ASL, unitamente alle copie delle prescrizioni medico-veterinarie di cui all'articolo 76, comma 1, ed alla documentazione di acquisto, e' conservato per 5 anni dall'ultima registrazione anche in caso di abbattimento degli animali prima della scadenza di tale periodo ed e' esibito a richiesta della ASL per i controlli."
Risposta:
Il legislatore evidentemente non si è posto il problema dell’impossibilità per un registro che a ritroso contenga registrazioni antecedenti ai 5 anni, dello scorporo di queste.
Il registro andrà dunque tenuto 5 anni dopo l’ultima registrazione e potrà contenere registrazioni antecedenti ai 5 anni.
Se per un qualsiasi motivo il registro viene chiuso prima di essere ultimato, le righe e le pagine bianche andranno barrati e farà fede la data dell’ultima registrazione.
Le ricette e i documenti commerciali (per le finalità sanitarie) invece possono essere tenuti per il solo periodo di tempo preciso dei cinque anni.
E’ evidente che un periodo di cinque sia ritenuto sufficiente a coprire le tutele volute dalla legge sia in termini di sanità animale che di salute pubblica.
Domanda nr. 313
Inserita il 31/03/2014
Domanda:
L’agenzia delle entrate mi chiede un registro carico e scarico dei vaccini per piccoli animali. E’ obbligatorio detenerlo?
Risposta:
Al fine di rispondere precisamente è bene chiarire come per la legge non esistono “piccoli animali” ma animali produttori di alimenti per l’uomo e animali non produttori di alimenti per l’uomo.
In questo contesto non sono previsti dalla attuale normativa in materia di medicinali veterinari (193, art. 15 del 158, RPV '54, gas anestetici, omeopatici, mangimi medicati), speciali registri su cui annotare il carico e/o scarico di prodotti immunizzanti (vaccini o sieri). I prodotti immunizzanti, vaccini o sieri, sono considerati alla stessa stregua degli altri medicinali veterinari (es. antibiotici, antinfiammatori, anti parassitari, ecc.).
Sono invece medicinali "speciali" il cui uso e detenzione abbisogna di speciale registro vidimato dall'autorità competente, alcune categorie di stupefacenti (art.42, L.309/90, Tab.II, sezione A,B,C) ed alcuni particolari ormoni e per certi usi (art.4, 5 D.Lgs.158/2006).
Per i medicinali per i quali non è previsto un registro specifico (es. vaccini, antibiotici, antinfiammatori, ecc.), quando usati, come nel caso qui proposto, in animali non produttori di alimenti per l'uomo (animali d'affezione) le operazioni di carico sono soddisfatte con la tenuta dei documenti commerciali di acquisto dei medesimi per un periodo di 3 anni, e lo scarico si effettua su un registro di scarico vidimato solo quando essi dovessero essere usati in animali produttori di alimenti per l'uomo (art. 84, comma 4, D.Lgs 193/2006).
La legge ad es. diversifica i conigli che possono essere sia DPA che non DPA ma non lo fa per molte specie animali che oggi si possono presentare in ambulatorio, essere dichiarate nella volontà del proprietario quali non-DPA ma dover, di fatto, essere trattate a tutti gli effetti come DPA dal veterinario nel rispetto della normativa come ad es. polli, maialini nani, capre e caprette, oche bianche.... ecc.
Domanda nr. 312
Inserita il 31/03/2014
Domanda:
Vorrei sapere se quanto sostenuto dalle FAQ 133, 126 e 6 sia cambiato qualcosa in merito al rifornimento dell’ossigeno ma anche dei gas anestetici e medicinali.
Risposta:
Data la ricorrenza della domanda e l’importanza del problema il GdL farmaco veterinario ha riesaminato tutta la legislazione e chiesto il parere del legale.
Il DLgs 193/06 all’art. 3 “Fattispecie escluse dalla disciplina” recita: 1. Il presente decreto non si applica: ... omissis..... f) ai gas anestetici ai quali si applica la disciplina di cui al
Tale Decreto infatti non esclude dal rifornimento i veterinari laddove per il rifornimento parla solo di “soggetti aventi diritto” senza mai vincolare la sua applicazione a medici piuttosto che veterinari.
E’ indubbio che il DLgs 219 sia normativa speciale nel regolamentare la fattispecie in quanto tratta a tutto tondo il tema dei gas medicinali e perciò prevalente rispetto al DLgs 193/06.
Per quanto attiene ai gas medicinali in generale è evidente come lo spirito della legge intendesse non vincolare l’esclusione dal campo di applicazione dei soli gas anestetici per i quali dunque valgono le considerazioni di cui sopra.
Le risposte alle FAQ citate si riferiscono tutte in realtà all’applicazione della norma dettata da una nota di chiarimento del Ministero relativa all’assimilazione, giustamente, dei gas medicali quali medicinali ad uso umano provvisti di AIC che, in assenza dell’esclusione dal campo di applicazione succitata, dovrebbero seguire per il rifornimento, l’iter indicato nella FAQ n° 6.
Per quanto riguarda dunque il rifornimento, ogniqualvolta si tratti di gas medicinali non anestetici, in attesa di chiarimenti, si consiglia tale iter.
Si ringrazia l'avv. Scarciglia per il contributo
Domanda nr. 311
Inserita il 24/03/2014
Domanda:
Dove sta scritto l’obbligo di vidimazione annuale del registro stupefacenti di un grossista di medicinali veterinari autorizzato anche alla vendita diretta?
Risposta:
L'articolo 62 del DPR 309/90 prevede che la chiusura del registro a fine anno si faccia mediante scritturazione riassuntiva dal parte del farmacista responsabile del registro.
Per cui non è prevista alcuna vidimazione per tale chiusura da parte di alcun Ente controllore.
Domanda nr. 310
Inserita il 24/03/2014
Domanda:
Confrontando la risposta data alla faq 303 e ciò che viene detto nella nota allegata, sembra esista una evidente contraddizione in merito alla possibilità per un medico veterinario zooiatra, senza ambulatorio, di potersi autoricettare con la triplice (RNRT) un farmaco uso umano in deroga ex art.11 - D.Lvo 193/2006 ad animale DPA. Ai sensi della medesima (nota) viene infatti negata tale possibilità, quindi niente autoprescrizione con la "triplice". Rimane dunque per un veterinario zooiatra la possibilità: o di ricettare direttamente l'"umano" al proprietario; oppure autoricettarsi un farmaco uso umano utilizzando altre tipologie di ricette, autointestandosele e scrivendo sopra autoprescrizione. La procedura per poter acquisire il farmaco umano sarebbe ad ogni buon conto questa visto che di autoprescrizione la norma non ne parla?
Risposta:
La risposta alla faq 303 datata 26/2/2014 si riferiva alla nota del Ministero emanata allora in risposta ad un quesito FNOVI (nota 11092/2011). In base a tale nota il veterinario zooiatra che operi su un animale DPA può auto prescriversi il farmaco ad uso umano con RNRT nel rispetto degli articoli 10 e 11. In data 2/2/2012 per la medesima situazione il Ministero nega tale possibilità.
Il problema non è la FAQ ma le due note ministeriali che sembrano contraddirsi.
Per quanto riguarda l’assenza di specifica dell’autoprescrizione nel DLgs 193/06 si precisa come la prescrizione sia una certificazione emessa dal veterinario che attesta che un certo soggetto ha diritto ad acquisire un farmaco e con cui ne autorizza l'acquisto. Senza tale autorizzazione tale soggetto non avrebbe diritto a detenere il farmaco in quanto la legge glielo vieterebbe (obbligo di prescrizione).
Se il soggetto che ha necessità e quindi diritto ad acquisire il medicinale é lo stesso veterinario prescrittore si ha un'autoprescrizione.
Il decreto legislativo 193 non specifica la cosa perché la questione superflua in quanto non esiste una legge (ci mancherebbe) che vieta di prescrivere a sé stessi.
Domanda nr. 309
Inserita il 20/03/2014
Domanda:
In relazione all’uso in deroga vorrei usare il principio attivo pentossifillina in un equide non DPA. Non mi risulta esistano farmaci veterinari in Italia contenenti tale principio attivo. Posso prescrivere al proprietario di un equide non DPA il farmaco TRENTAL® 30 cpr 400mg ad uso umano a base di pentossifillina? Se si, posso prescriverlo in ricetta unica ripetibile?
Risposta:
La normativa impone di utilizzare, in via prioritaria i farmaci registrati per una data patologia della la specie trattata.
Il problema non è sapere “quale principio attivo utilizzare” ma sapere “per quale patologia è registrato quel principio attivo in un medicinale”. Si tratterà allora di vedere, se per quella patologia e per quella specie siano registrati altri farmaci, indipendentemente dal principio attivo ivi contenuto.
Se la risposta è si, si dovrà usare quel farmaco, se la risposta è no, si procederà nel rispetto dell’uso a cascata così come regolamentato , in questo caso, per equidi non-DPA.
Domanda nr. 308
Inserita il 20/03/2014
Domanda:
Si chiede se un veterinario aziendale in una società agricola quale dipendente senza partita IVA possa ricettare e tenere il registro dei trattamenti e/o scorte in alcuni allevamenti in soccida con il proprio titolare. Il registro è intestato al soccidario, mentre il soccidante (il titolare) è proprietario degli animali.
Risposta:
Nulla osta ai sensi dell’art. 81 del DLgs 193/06 a che in queste condizioni il medico veterinario possa essere responsabile dei trattamenti e della scorta.
Come in tutte le aziende sarà necessario chiarire chi sia il responsabile dei trattamenti farmacologici e della scorta: il soccidante o il soccidario.
Domanda nr. 307
Inserita il 11/03/2014
Domanda:
E' legittima la cessione/vendita da parte dei medici veterinari dei prodotti per la prevenzione della filariosi cardiopolmonare durante l'attività clinica ambulatoriale?
Risposta:
Il testo di legge e il chiarimento non lasciano spazio a dubbi in merito al fatto che la cessione del farmaco sia una prestazione accessoria concessa a raffronto di una diagnosi che giustifichi la terapia. La prestazione accessoria "cessione" necessita, a monte, di un'altra prestazione principale la cui natura abbia attinenza col farmaco da cedere. Nel caso specifico a raffronto di un test annuale di controllo per la filaria, la cessione del farmaco rientra in questa fattispecie anche in assenza di malattia conclamata in quanto terapia di una fase embrionale. In ogni caso, come si evince dal foglietto illustrativo, la specialità medicinale ha come indicazione la prevenzione della filariosi cardio-polmonare, tuttavia si configura come trattamento a tutti gli effetti in caso di infestazione di microfilarie peraltro di difficile individuazione in taluni casi o per infestazione lieve).
La terapia con Cardotek® , e con altri medicinali analoghi, è infatti una terapia profilattica finalizzata ad uccidere le microfilarie per evitare che diventino vermi adulti. Questi farmaci trovano la loro indicazione a patologia già in atto, anche se allo stadio embrionale (larvale) e non rientrano tra i medicinali che si somministrano per evitare che la malattia venga contratta come nel caso di un vaccino. Diventa difficile ipotizzare di contestare la vendita del Cardotek® ad un professionista che abbia fatto una diagnosi secondo cui serviva quel farmaco. La legge infatti non vincola la cessione al rispetto dell'AIC ma al rispetto di un bisogno terapeutico. Per completezza si allega la Nota del Ministero della Salute 23711 del 24/12/2012 inerente la cessione e la registrazione dell'eventuale scarico dalla scorta di farmaci del Medico Veterinario o della Struttura.
Risposta aggiornata al 13/11/2019
Domanda nr. 306
Inserita il 10/03/2014
Domanda:
In merito alle ricette degli allevamenti intensivi di conigli e suini contenenti prescrizioni di medicinali prefabbricati e di additivi da usare per via orale o da miscelare nei mangimi o sfarinati e di antibiotici da usare per via orale, chiedo se questi allevamenti debbono avere regolare autorizzazione Ministeriale circa le operazioni di miscelazione al fine di garantire l'idonea omogeneità del prodotto finito.
Risposta:
Premesso che i medicinali veterinari prefabbricati non esistono più e che mangimi o sfarinati sono sinonimi, la prescrizione di medicinali veterinari da somministrarsi in alimenti liquidi o solidi, nonché in acqua di bevanda (verificando se l'AIC li contempla), non richiede alcun tipo di autorizzazione, tranne che per le premiscele medicate (che sono sempre e comunque medicinali veterinari), che possono essere prescritte solo a quegli allevamenti che sono autorizzati dal Ministero della Salute congiuntamente al Ministero dell'Industria-commercio-artigianato a produrre mangimi medicati per gli animali allevati.
A completamento della risposta si sottolinea come nell'attività di farmacosorveglianza, vada posta particolare attenzione alla verifica delle "attrezzature" presenti in allevamento atte a consentire la somministrazione di tali trattamenti.
Ad esempio, per un trattamento in acqua di bevanda, va verificata sempre la presenza di apparecchiature in grado di diluire il medicinale (Dosatron o altro) e questo non in virtù dello stretto dettame normativo ma in virtù di una prassi che, come controllori, è dovuta, ossia quella di verificare per un uso proprio del medicinale anche il rispetto delle modalità della via di somministrazione oltre alla via di somministrazione stessa.
Domanda nr. 305
Inserita il 10/03/2014
Domanda:
Un allevamento ovino con annesso caseificio e laboratorio sezionamento carne autorizzato alla detenzione di scorte di farmaci veterinari intende detenere e somministrare alcuni farmaci veterinari contenenti sostanze di cui agli articoli 4 e 5 del D.L vo 158/2006 per sincronizzare gli estri delle femmine del gruppo; nel caso specifico il "Crono-gest ® (p.a. cronolone) e "CIDR 1,38 ® p.a. progesterone). Nella FAQ 36 era esplicitata la possibilità di ricettare il CIDR® in un allevamento con scorta e la compilazione del relativo registro. Le recenti Linee guida del 158/2006 del 04/03/2013, nel paragrafo 3, citano "Si ribadisce che presso le aziende, anche se autorizzate ai sensi dell'articolo 80 del D.L. vo 193/2006, non potranno essere detenute scorte di tali medicinali veterinari". Tenuto conto della necessità di mantenere sempre la tracciabilità dei trattamenti e dall'altro le formulazioni delle confezioni, talvolta multiple, qual'è il comportamento più corretto : approvvigionamento limitato al necessario con prescrizione RNTC ad animali identificati (tatuaggio/bolo, etc..) e successiva registrazione delle somministrazioni nel registro rosa con tutte le indicazioni previste dall'articolo 4 del D.L. vo 158/2006?
Risposta:
Come altre volte in queste FAQ, si premette che le note ministeriali al pari delle linee guida non hanno valore di legge laddove creano diritto normativo novativo inteso sia quale nuovo dettame che come deroga.
Per la risposta generale in merito alla detenzione di queste sostanze si veda la FAQ 36 che esplicita come ai sensi dell'art 4 d.l. 158, a fini terapeutici, nulla vieta la detenzione anche in scorta di tali medicinali, se si tratta di allevamento da riproduzione e non da ingrasso.
Questo impianto normativo viene confermato dal dettame del DM 28 luglio 2009.
Per la particolare situazione descritta tuttavia si tenga presente come il far riferimento ad un allevamento con annesso caseificio e laboratorio sezionamento carne evidenzia come questo non sia solo un allevamento da riproduzione, pertanto la detenzione in scorta è vietata.
Notare che la norma fa una esplicita distinzione, con due diversi articoli, 4 e 5 tra trattamento terapeutico e trattamento zootecnico Pertanto la detenzione è vietata anche perchè si prevede l'uso del medicinale per trattamento zootecnico (sincronizzazione degli estri) e per tale utilizzo, l'art.5 comma 2 d.l. 158 prevede la compilazione di una ricetta non di scorta.
E’ dunque corretto il comportamento descritto nel quesito.
Domanda nr. 304
Inserita il 04/03/2014
Domanda:
Con la presente sono a porre una domanda tecnica. Se in una struttura veterinaria viene effettuata la terapia con la talidomide, preparata dal farmacista, tale preparazione deve essere riportata nel registro stupefacenti, oppure serve un altro apposito registro?
Risposta:
La Talidomide non è uno stupefacente, anche se sedativo ipnotico.
E’ un principio attivo non compreso nella tabella II sez. A, B, e C della 309/90, e non deve dunque essere registrato nel registro degli stupefacenti.
Non potendo essere somministrata ad animali DPA nessun altro registro è necessario per il suo utilizzo in animali non-DPA
Domanda nr. 303
Inserita il 26/02/2014
Domanda:
Nel caso in cui un veterinario zooiatra voglia approvvigionarsi di un farmaco uso umano per uso contingente mediante autoricettazione, deve utilzzare la ricetta ministeriale in triplice copia o va bene anche la ricetta su carta intestata?
Risposta:
La RNRTC é il modello obbligatorio per l'approvvigionamento per scorta dei soggetti di cui all'art. 65. In tutti gli altri casi, e quindi anche per l'autoprescrizione
Nel caso dunque di medicinali per uso umano da somministrare ad un animale DPA in applicazione dell’uso in deroga di cui all’art. 11 del DLgs 193/06, andranno prescritti con RNRT come da modello All.III del 193.
Andranno invece prescritti con ricetta non ripetibile se utilizzati in deroga in applicazione dell’art. 10, su animali non-DPA.
Domanda nr. 302
Inserita il 18/02/2014
Domanda:
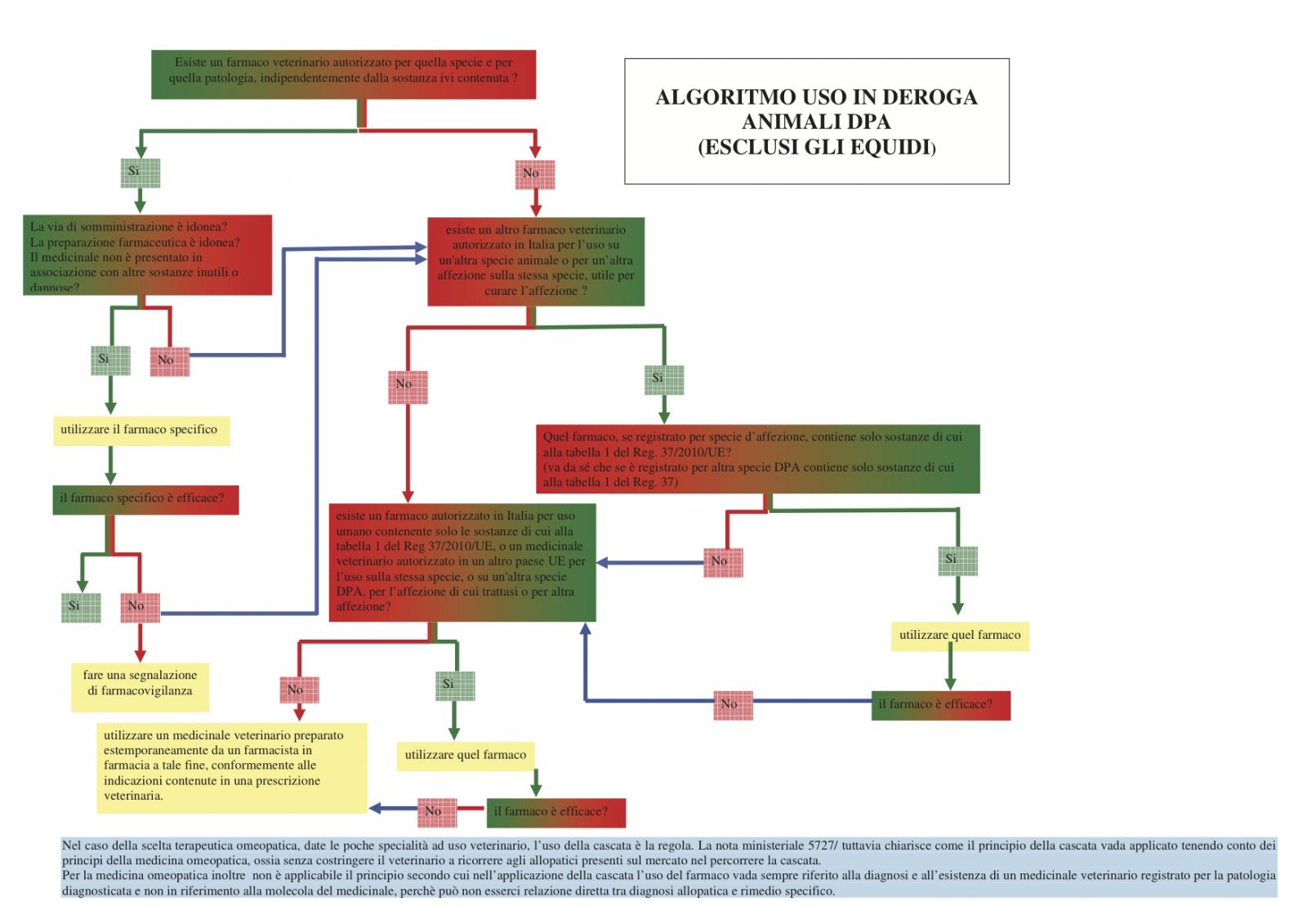
Esiste un diagramma di flusso che comprenda tutte le regole dell’uso a cascata (in deroga) del farmaco negli animali non-DPA?
Risposta:
Si veda anche la domanda n.268.
Domanda nr. 301
Inserita il 18/02/2014
Domanda:
Esiste un diagramma di flusso che comprenda tutte le regole dell’uso a cascata (in deroga) del farmaco negli animali DPA?
Risposta:
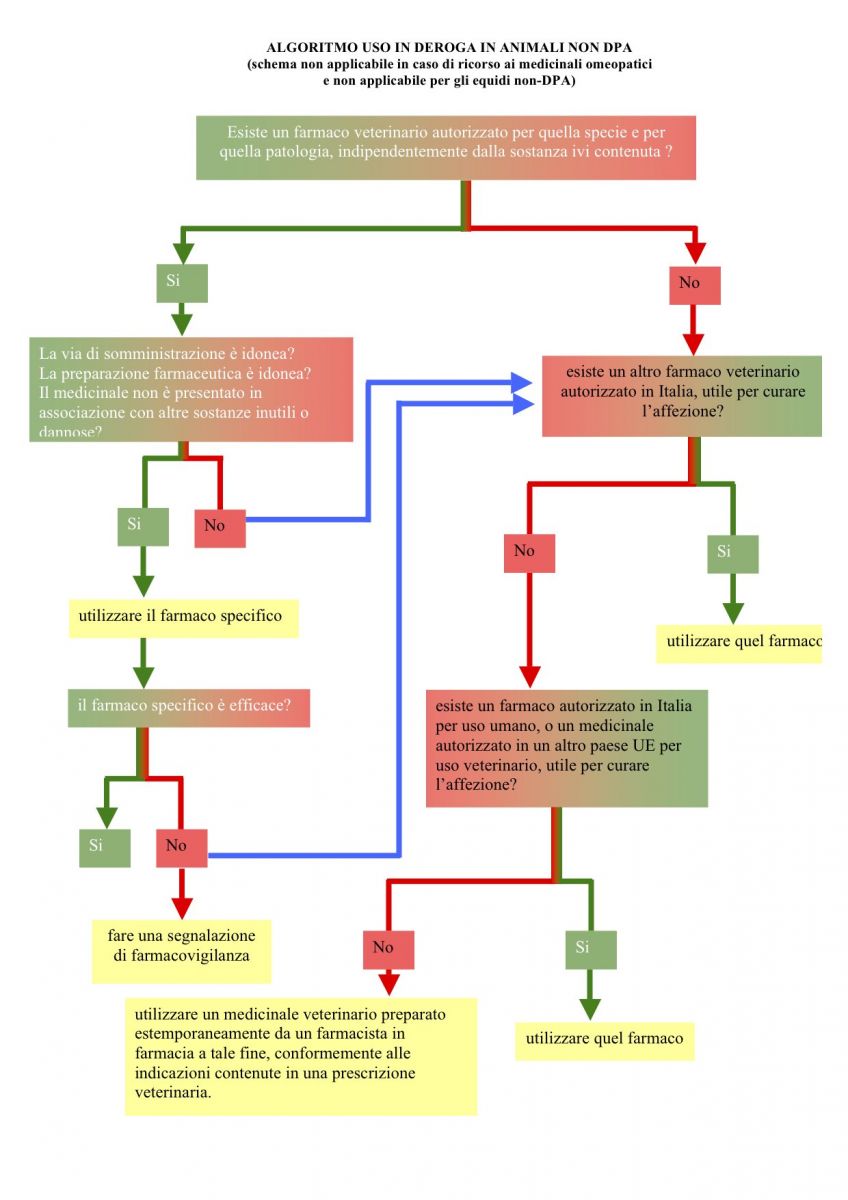
Domanda nr. 300
Inserita il 17/02/2014
Domanda:
In merito alla gestione e detenzione delle scorte zooiatriche vorrei sapere se nel caso di un medico veterinario dotato di autorizzazione alla detenzione di scorte zooiatriche che si occupa di bovini e cavalli (DPA e non DPA) gli obblighi relativi al carico e scarico dei farmaci veterinari siano i seguenti:
- Carico dei farmaci veterinari: viene assolto conservando la copia gialla della ricetta e il documento di trasporto/ fattura immediata;
- Scarico dei farmaci delle proprie scorte: o se l’animale è un bovino o un cavallo DPA bisogna registrare sul proprio registro e sul registro aziendale delle somministrazioni dei farmaci lo scarico del farmaco; o se l’animale è un cavallo non DPA è sufficiente indicare il farmaco utilizzato indicandolo come voce a parte in fattura.
Risposta:
In linea di massima le considerazioni sono esatte salvo che nulla è dovuto sulla fattura.
Lo scarico sul registro della scorta zooiatrica o dell'impianto in cui vengono curati o custoditi animali avviene quando tali medicinali sono stati somministrati ad animali DPA A giustificativo del carico deve essere conservata anche la copia della ricetta Inoltre ai proprietari degli animali nDPA possono essere cedute le confezioni di medicinali veterinari della scorta zooiatrica allo scopo di iniziare la terapia (art.84 comma 3 D.L.gs 193/06).
Per quanto riguarda le modalità di registrazione si segnala la nota del MinSal 2709 del 12/2/2013.
Domanda nr. 299
Inserita il 17/02/2014
Domanda:
E’ possibile prescrivere il Danilon® ad un cavallo DPA? Non sono riuscita a trovarlo né nel Reg. 37 né nel Reg. 1950. Il principio attivo è il suxibuzone, è un FANS.
Risposta:
Gli equidi DPA nel rispetto della cascata ossia in assenza di un farmaco registrato per loro per la patologia specifica, per le motivazioni di cui all’articolo 11 del DLgs 193/06, possono accedere a farmaci registrati per altre specie animali o per l’uomo a condizione che le molecole siano contenute o nella tabella 1 del reg. 37/2010/UE o nel Regolamento 1950/2006/CE.
I farmaci che contengono Suxibuzone, non essendo questo principio attivo presente in nessuno dei due elenchi, non potranno essere somministrati agli equidi DPA.
Domanda nr. 298
Inserita il 12/02/2014
Domanda:
Qual è la corretta tenuta del registro di carico-scarico dei farmaci nella gestione di un ambulatorio veterinario? Ho notato che in alcune Vs risposte ad altre domande sullo stesso argomento avete specificato il fatto di avere la degenza di animali d'affezione.
Risposta:
Le modalità di registrazione sono quelle previste all'art. 84 del D. Leg. 193/2006: il carico viene assolto con la conservazione della documentazione d'acquisto per 3 anni. Lo scarico deve essere effettuato, entro 7 gg lavorativi, solo in caso di somministrazione di medicinali ad animali DPA.
In quest'ultimo caso il registro deve essere conservato per almeno 5 anni dall'ultima annotazione.
Domanda nr. 297
Inserita il 12/02/2014
Domanda:
L'approvvigionamento di farmaci ospedalieri (esclusi gli antibatterici) da parte di una clinica veterinaria, è possibile direttamente dalla ditta produttrice senza passare da un fornitore?
Nel caso in cui fosse possibile, potete mandarmi il riferimento legislativo?
Risposta:
Per i farmaci ospedalieri (esclusi gli antibatterici) la risposta è affermativa e il riferimento legislativo è l’art. l?84 comma 7.
Per l’approvvigionamento invece dei farmaci prescrivibili solo da uno specialista, questi potranno essere acquistati solo nelle farmacie aperte al pubblico.
Domanda nr. 296
Inserita il 12/02/2014
Domanda:
Si chiede di sapere se un veterinario possa prescrivere, con ricetta in triplice copia, il farmaco ZOLETIL 100 (anestetico) ad un proprietario di cani. Nel caso non fosse possibile si chiede di sapere a quale sanzione va incontro il veterinario prescrittore, il farmacista che ha venduto il farmaco e il proprietario dei cani che ha usato il farmaco.
Risposta:
Lo Zoletil é un anestetico generale. Pertanto rientra tra i medicinali di cui, ai sensi del DM 28 luglio 2009, l'uso e la detenzione sono riservati al veterinario.
Per quanto riguarda le sanzioni:
- 108 comma 9 per veterinario, farmacista e proprietario dato che il comma nove recita ?chiunque non osserva? (da euro 2.582,00 a euro 15.493,00)
- in caso di utilizzo del farmaco per il proprietario si potrebbe configurare l’abuso di professione, perseguito dal codice penale.
Domanda nr. 295
Inserita il 12/02/2014
Domanda:
Quale articolo di legge viene violato quando un veterinario prescrive un medicinale con dosaggi e/o durata di trattamento diversi da quelli del foglietto illustrativo (AIC) quali ad es dosaggi decuplicati o prescritti in one-shot a fronte dell'AIC che prevede una durata di trattamento almeno di 3-5giorni ? Si precisa che specie e patologia sono quelle del foglietto illustrativo.
La relativa sanzione è chiara ed è stata ribadita recentemente dal Ministero nelle varie note e linee guida (trattasi della sanzione di cui all'art.108, comma 9 del 193/06)
Risposta:
Nel caso descritto patologia e specie di destinazione corrispondendo a quanto previsto dall’AIC, non si tratta di uso in deroga in quanto il veterinario non si ritrova nella condizione di assenza di farmaco ma di un suo utilizzo in difformità da quanto indicato nel foglietto illustrativo.
L’articolo disatteso è il 9 del DLgs 193/06 in riferimento alla definizione di autorizzazione? di cui all’art. 5 che indica chiaramente come la somministrazione ammessa è quella che rispetta l’autorizzazione comprensiva del dosaggio del farmaco. La sanzione è quella del comma 9 dell’art. 108 che recita: “Salvo che il fatto costituisca reato, chiunque non osserva le prescrizioni imposte con le autorizzazioni rilasciate a norma del presente decreto è soggetto al pagamento di una sanzione amministrativa pecuniaria da euro 2.582,00 a euro 15.493,00”.
Domanda nr. 294
Inserita il 05/02/2014
Domanda:
Si chiede, alla luce della nota allegata (seppur datata) e dell'attuale normativa, quali siano gli obblighi per allevatori di animali DPA riguardanti l'acquisto, detenzione e somministrazione di medicinali veterinari dispensabili senza ricetta, non avendo la suddetta nota minimamente richiamato i medesimi (obblighi).
Risposta:
In merito al quesito è necessario chiarire come:
1) la legislazione sia chiara e imponga la registrazione di tutti i trattamenti da parte dell’allevatore
2) la nota allegata ribadisce questo concetto pur sostenendo che il rinnovo della normativa dovrebbe valutare di modificare questo stato di cose
3) diverse note ministeriali sono sopravvenute a ribadire ma anche a contraddire la legge in merito alle registrazioni dovute per farmaci prescritti con ricette diverse dalla RNRT (13/5/2009 n° 8988 - nota del 15/9/2009 n° 16361)
4) l’ultima, la nota 7835 del 4/3/2013 ribadisce quanto sostenuto in una delle precedenti ossia che: “Si precisa che, nel registro dei trattamenti, i medicinali veterinari dispensabili con prescrizione in triplice copia sono registrati con le modalità previste dall’art. 79 del decreto legislativo n. 193 del 2006. Per quanto riguarda i farmaci dispensabili mediante altre tipologie di prescrizione, vista da un lato l’inderogabile necessità di garantire la tracciabilità del farmaco veterinario e la terapia effettuata e dall'altro l’opportunità di consentire lo snellimento delle procedure burocratiche di registrazione dei medicinali veterinari per i quali non sono previsti tempi di sospensione, gli adempimenti di registrazione si ritengono assolti mediante la conservazione delle ricette medesime per un periodo di almeno 5 anni e la registrazione, nel registro dei trattamenti, del numero della ricetta, della data di emissione e della data di inizio del trattamento.”
LINEE GUIDA APPLICATIVA del Decreto Legislativo del 16 marzo 2006, n. 158 per l’armonizzazione dei controlli ufficiali volti alla ricerca di residui di sostanze chimiche potenzialmente pericolose durante il processo di allevamento e di prima trasformazione dei prodotti di origine animale, ai sensi del decreto legislativo 16 marzo 2006, n.158 e successive modifiche.
Tralasciando il fatto che è impossibile trattenere la copia che non esiste della RNR, non chiarendo ancora il Ministero della Salute quale sia il comportamento per i farmaci dispensabili senza ricetta, si arriverebbe all’assurdo che per questi tutte le registrazioni siano dovute diversamente da quelli dispensabili con RNR e RR.
Alla luce di quanto sopra, e considerato che una nota ministeriale non può derogare da un dettame di legge, l’unico dettame certo è quello della normativa, all’art. 79 del DLgs 193/06 e che, come si può leggere, non distingue ne tra le varie tipologie di prescrizione ne tra farmaci dispensabili con o senza ricetta.
Domanda nr. 293
Inserita il 05/02/2014
Domanda:
Nel caso un medico veterinario sia titolare di due cliniche per animali da affezione in due regioni diverse, ma solo in una clinica sia anche direttore sanitario, può fare degli interventi nella clinica dove non è direttore sanitario utilizzando medicinali prelevati dalla scorta dell'altra clinica dove lui ne è il responsabile ?
Risposta:
No perché la scorta é riferita ad una struttura specifica.
La cosa peraltro trova conferma indiretta nella disposizione dell'art. 84 del D. Leg. 193/2006 che prevede che le scorte dell'impianto di cura non possano essere utilizzate neanche per le visite domiciliari a meno che non abbiano carattere di urgenza.
Domanda nr. 292
Inserita il 05/02/2014
Domanda:
Il medico veterinario che accetta di essere responsabile delle scorte di un canile (rifugio) art 82 D.Lvo 193/2006 è tenuto a dichiarare nella accettazione di essere già responsabile di scorta di farmaci presso altri impianti ? L'eventuale omissione è sanzionabile ?
Risposta:
Se la scorta è per un impianto di cura di animali, non serve indicare se si è responsabili di altre scorte (art.84, 193/06), qui il responsabile è il direttore sanitario della struttura.
Se la scorta è per un impianto di allevamento e custodia di animali NON DPA, si, bisogna indicare se si è veterinari responsabili di altre scorte (art. 82, 193/06).
La scorta di un canile rifugio come indicata dal quesito, rientra nell'art. 82 e/o contemporaneamente negli artt. 82 e 84, per cui la risposta è sempre si, dovrà essere indicata l'eventuale responsabilità su altre scorte, in quanto un rifugio è sempre o anche per custodia.
Per quanto riguarda la sanzione: - se l'indicazione non é inserita nella richiesta di autorizzazione, non viene data l'autorizzazione (o viene ritirata in autotutela se é stata erroneamente concessa) ma non c'é sanzione; - se viene inserita un'indicazione falsa il reato è penale.
Domanda nr. 291
Inserita il 29/01/2014
Domanda:
Come devono essere fatte le correzioni sul registro degli stupefacenti?
Risposta:
La correzione deve essere apportata barrando con una riga la parte errata, in modo che continui ad essere visibile, e scrivendo a fianco il dato corretto, nonché datando e firmando la correzione nei pressi della stessa.